IntaRNA 

Efficient RNA-RNA interaction prediction incorporating accessibility and seeding of interaction sites
During the last few years, several new small regulatory RNAs (sRNAs) have been discovered in bacteria. Most of them act as post-transcriptional regulators by base pairing to a target mRNA, causing translational repressionex or activation, or mRNA degradation. Numerous sRNAs have already been identified, but the number of experimentally verified targets is considerably lower. Consequently, computational target prediction is in great demand. Many existing target prediction programs neglect the accessibility of target sites and the existence of a seed, while other approaches are either specialized to certain types of RNAs or too slow for genome-wide searches.
IntaRNA, developed by Prof. Backofen’s bioinformatics group at Freiburg University, is a general and fast approach to the prediction of RNA-RNA interactions incorporating both the accessibility of interacting sites as well as the existence of a user-definable seed interaction. We successfully applied IntaRNA to the prediction of bacterial sRNA targets and determined the exact locations of the interactions with a higher accuracy than competing programs.
For testing or ad hoc use of IntaRNA, you can use its webinterface at the
==> Freiburg RNA tools IntaRNA webserver <==
Contribution
Feel free to contribute to this project by writing Issues with feature requests, bug reports, or just contact messages.
Citation
If you use IntaRNA, please cite our respective articles
Method
- IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions Martin Mann, Patrick R. Wright, and Rolf Backofen, Nucleic Acids Research, 45 (W1), W435–W439, 2017, DOI:10.1093/nar/gkx279.
- IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions Anke Busch, Andreas S. Richter, and Rolf Backofen, Bioinformatics, 24 no. 24 pp. 2849-56, 2008, DOI:10.1093/bioinformatics/btn544.
Features and Application
- Integration of accessibility data from structure probing into RNA–RNA interaction prediction Milad Miladi, Soheila Montaseri, Rolf Backofen, Martin Raden, Bioinformatics, 2019, DOI:10.1093/bioinformatics/bty1029.
- IntaRNAhelix - Composing RNA-RNA interactions from stable inter-molecular helices boosts bacterial sRNA target prediction Rick Gelhausen, Sebastian Will, Ivo L. Hofacker, Rolf Backofen, and Martin Raden, Journal of Bioinformatics and Computational Biology, 2019, 17(5), 1940009, DOI:10.1142/S0219720019400092.
- CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains Patrick R. Wright, Jens Georg, Martin Mann, Dragos A. Sorescu, Andreas S. Richter, Steffen Lott, Robert Kleinkauf, Wolfgang R. Hess, and Rolf Backofen, Nucleic Acids Research, 42 (W1), W119-W123, 2014, DOI:10.1093/nar/gku359.
- The impact of various seed, accessibility and interaction constraints on sRNA target prediction- a systematic assessment. Martin Raden, Teresa Müller, Stefan Mautner, Rick Gelhausen, and Rolf Backofen, BMC Bioinformatics, 21 (15), 2020, DOI:10.1186/s12859-019-3143-4.
Documentation
Overview
The following topics are covered by this documentation:
- Installation
- Usage and Parameters
- Just run …
- General things you should know
- Interaction Model
- Prediction modes
- IntaRNA’s multiple personalities
- IntaRNA - fast, heuristic RNA-RNA interaction prediction
- IntaRNAhelix - helix-based predictions
- IntaRNAexact - exact predictions like RNAup
- IntaRNAduplex - hybrid-only optimization like RNAduplex
- IntaRNAsTar - optimized for sRNA-target prediction
- IntaRNAseed - identifys and reports seed interactions only
- IntaRNAens - ensemble-based prediction and partition function computation
- How to constrain predicted interactions
- Output Setup
- Output modes
- Pairwise vs. all-vs-all
- Sequence indexing
- Suboptimal RNA-RNA interaction prediction and output restrictions
- Energy parameters and temperature
- Additional output files
- Library for integration in external tools
- Auxiliary R scripts for output visualization etc.
- Auxiliary python scripts for IntaRNA-based pipelines
Installation
IntaRNA via conda (bioconda channel)
The most easy way to locally install IntaRNA is via conda using the
bioconda
channel (linux only). This way, you will install a pre-built IntaRNA binary along
with all dependencies.
Follow
![]() to get detailed information or run the following command to install it to the currently activated environment.
to get detailed information or run the following command to install it to the currently activated environment.
conda install -c conda-forge -c bioconda intarna
Note: Conda is available for Windows, MacOs and Linux. We recommend the Miniconda installation
Note further: Windows user without Powershell or Commandline experience might consider to first install a Linux App via WSL and use and install conda therein since most related scripts will be tailored for Linux BASH shells or similar systems.
IntaRNA docker container (via QUAY.IO)
An IntaRNA docker container (?) is provided from the bioconda package via Quay.io. This gives you with an encapsulated IntaRNA installation.
Note The biocontainer builds do not support the latest tag, such that you have to specify a version to download! So best
- open the quay.io IntaRNA tag page
- identify the tag of the container version you want to install, e.g.
3.4.1--pl5321h077b44d_3 - use this tag in your container pull command as an appendix, e.g.
podman pull quay.io/biocontainers/intarna:3.4.1--pl5321h077b44d_3ordocker pull quay.io/biocontainers/intarna:3.4.1--pl5321h077b44d_3
Dependencies
If you are going to compile IntaRNA from source, ensure you meet the following dependencies:
- compiler supporting C++11 standard and OpenMP
- boost C++ library version >= 1.50.0
(ensure the following libraries are installed for development (not just runtime libraries!); or install all e.g. in Ubuntu via package
libboost-all-dev)- libboost_regex
- libboost_program_options
- libboost_filesystem
- libboost_system
- Vienna RNA package version >= 2.4.14
pkg-configfor detailed version checks of dependencies- if cloning from github: GNU autotools (automake, autoconf, ..)
Also used by IntaRNA, but already part of the source code distribution (and thus not needed to be installed separately):
- Catch test framework
- Easylogging++ logging framework
Cloning Source code from github (or downloading ZIP-file)
The data provided within the github repository
(or within the Source code archives provided at the
IntaRNA release page)
is no complete distribution and
lacks all system specifically generated files. Thus, in order to get started with
a fresh clone of the IntaRNA source code repository you have to run the GNU autotools
to generate all needed files for a proper configure and make. To this end,
we provide the helper script autotools-init.sh that can be run as shown in the following.
# call aclocal, automake, autoconf
bash ./autotools-init.sh
Afterwards, you can continue as if you would have downloaded an IntaRNA package distribution.
IntaRNA package distribution (e.g. intaRNA-2.0.0.tar.gz)
When downloading an IntaRNA package distribution (e.g. intaRNA-2.0.0.tar.gz) from the
IntaRNA release page, you should
first ensure, that you have all dependencies installed. If so, you can
simply run the following (assuming bash shell).
# generate system specific files (use -h for options)
./configure
# compile IntaRNA from source
make
# run tests to ensure all went fine
make tests
# install (use 'configure --prefix=XXX' to change default install directory)
make install
# (alternatively) install to directory XYZ
make install prefix=XYZ
If you installed one of the dependencies in a non-standard directory, you have
to use the according configure options:
--with-vrna: the prefix where the Vienna RNA package is installed--with-boost: the prefix where the boost library is installed
Note, the latter is for instance the case if your configure call returns an
error message as follows:
checking whether the Boost::System library is available... yes
configure: error: Could not find a version of the library!
In that case your boost libraries are most likely installed to a non-standard
directory that you have to specify either using --with-boost or just the
library directory via --with-boost-libdir.
Microsoft Windows installation
… from source
IntaRNA can be compiled, installed, and used on a Microsoft Windows system when e.g. using Cygwin as ‘linux emulator’. Just install Cygwin with the following packages:
- Devel:
- make
- gcc-g++
- autoconf
- automake
- pkg-config
- Libs:
- libboost-devel
- Perl:
- perl
and follow either install from github or install from package.
Note, the source code comes without any waranties or what-so-ever (see licence information)!
… using pre-compiled binaries
For some releases, we also provide precompiled binary packages for Microsoft Windows at the IntaRNA release page that enable ‘out-of-the-box’ usage. If you want to use them:
- download the according ZIP archive and extract
- open a Windows command prompt
- run IntaRNA
Note, these binaries come without any waranties, support or what-so-ever! They are just an offer due to user requests.
If you do not want to work within the IntaRNA directory or don’t want to provide
the full installation path with every IntaRNA call, you should add the installation
directory to your Path System variable
(using a semicolon ; separator).
OS X installation with homebrew (thanks to Lars Barquist)
If you do not want to or can use the pre-compiled binaries for OS X available from
bioconda, you can compile IntaRNA
locally.
The following wraps up how to build IntaRNA-2.0.2 under OS X (Sierra 10.12.4) using homebrew.
First, install homebrew! :)
brew install gcc --without-multilib
--without-multilib is necessary for OpenMP multithreading support – note
OS X default gcc/clang doesn’t support OpenMP, so we need to install standard
gcc/g++
brew install boost --cc=gcc-6
--cc=gcc-6 is necessary to build boost with standard gcc, rather than the
default bottle which appears to have been built with the system clang.
Brew installs gcc/g++ as /usr/local/bin/gcc-VERSION by default to avoid
clashing with the system’s gcc/clang. 6 is the current version as of
writing, but may change.
brew install viennarna
brew install doxygen
Download and extract the IntaRNA source code package (e.g. intaRNA-2.0.2.tar.gz) from the release page.
./configure CC=gcc-6 CXX=g++-6
This sets up makefiles to use standard gcc/g++ from brew, which will
need an update to the appropriate compiler version if not still 6.
You might also want to
set --prefix=INSTALLPATH if you dont want to install IntaRNA globally.
make
make tests
make install
Usage and parameters
IntaRNA comes with a vast variety of ways to tune or enhance YOUR RNA-RNA prediction. To this end, different prediction modes and interaction models are implemented that allow to balance predication quality and runtime requirement. Furthermore, it is possible to define interaction restrictions, seed constraints, explicit seed information, SHAPE reactivity constraints, output modes, suboptimal enumeration, energy parameters, temperature, and the accessibility handling. If you are doing high-throughput computations, you might also want to consider multi-threading support.
For ad hoc usage you can use the Freiburg RNA tools IntaRNA webserver (with limited parameterization).
Just run …
If you just want to start and are fine with the default parameters set,
you only have to provide two RNA sequences,
a (long) target RNA (using -t or --target) and a (short) query RNA
(via -q or --query), in
IUPAC RNA encoding.
You can either directly input the sequences
# running IntaRNA with direct sequence input
# call : IntaRNA -t CCCCCCCCGGGGGGGGGGGGGG -q AAAACCCCCCCUUUU
target
9 15
| |
5'-CCCCCCCC GGGGGGG-3'
GGGGGGG
+++++++
CCCCCCC
3'-UUUU AAAA-5'
| |
11 5
query
interaction energy = -10.85 kcal/mol
In case you need specific RNA names in your output, you can provide ID strings
for each RNA using e.g. --tId="mRNA with GC" or --qId="sRNA-example".
Multiple sequences can be provided in FASTA-format.
It is possible to use either file input or to read the FASTA input from the
STDIN stream.
# running IntaRNA with FASTA files
IntaRNA -t myTargets.fasta -q myQueries.fasta
# reading query FASTA input from stream via pipe
cat myQueries.fasta | IntaRNA -q STDIN -t myTargets.fasta
If you are working with large FASTA input files, e.g. covering a whole
transcriptome, you can restrict the prediction to a subset of the input
sequences using the --qSet or --tSet parameter as shown in the following.
# restrict prediction to the second load of 100 target sequences
IntaRNA -t myTranscriptome.fasta --tSet=101-200 -q myQuery.fasta
Furthermore, also gzip-compressed file input is supported and automatically
decompressed if the file name ends in .gz.
Nucleotide encodings different from ACGUT are rewritten as N and the respective
positions are not considered to form base pairs (and thus ignored).
Thymine T encodings are replaced by uracil U, since we apply an RNA-only
energy model.
For a list of general program argument run -h or --help. For a complete
list covering also more sophisticated options, run --fullhelp.
Multi-threading and parallelized computation
IntaRNA supports the parallelization of the target-query-combination processing.
The maximal number of threads to be used can be specified using the --threads parameter.
If --threads=k != 1, than k predictions are processed in parallel. A value of
0 requests the maximally available number of threads for this machine.
When using parallelization, you should have the following in mind:
-
The memory consumption will be multiplied by the number of threads, since each thread runs an independent prediction (with according memory consumption). Thus, ensure you have enough RAM available when using many threads of memory-demanding prediction modes. You might consider window-based prediction to limit the required RAM.
-
Parallelization is enabled hierarchically, ie. only one of the following input sets is processed in parallel:
- target sequences (if more than one)
- if only one target: query sequences (if more than one)
- if only one target and query: window combinations (if enabled)
The support for multi-threading can be completely disabled before compilation
using configure --disable-multithreading.
Load arguments from file
If you are using IntaRNA with similar command line arguments (parameters), you might want to reduce the call via the definition of a parameter file. To this end prepare a file that defines the common parameters in a the following form
#################################################################
# my parameter.cfg file to simulate RNAup predictions
#################################################################
# slow but exact predictions
mode = M
# no seed constraint
noSeed = true
# full global accessibility computation
accW = 0
accL = 0
where you use the long parameter names without the leading --.
Boolean arguments (like --noSeed) have to be set to true|1 or false|0
to be enabled or disabled, respectively. Given this,
you only have to pass the file name via --parameterFile=... and IntaRNA will
parse the additional parameters from your file.
Note: parameters specified via the command line take precedence over arguments from a parameter file. Thus, you can (silently) overwrite parameters that you have specified within the file.
Note further: parameter parsing from parameter file is (in contrast to the command line parsing) case sensitive!
General things you should know
RNA-RNA interaction models
IntaRNA supports various models how RNA-RNA interactions are represented. The model selection has direct consequences for the interaction patterns that can be predicted by IntaRNA. Before elaborating the supported models, first terms needed for understanding and representation:
We denote with a single site an interaction pattern of two respective RNA subsequences Qi..Qk and Tj..Tl that
- form a base pair on each end, i.e. (Qi,Tl) and (Qk,Tj) are pairing, and
- there are no intra-molecular base pairs within the two subsequences, i.e. the subsequences form only inter-molecular base pairs.
Given that we can classify single-site RNA-RNA interactions based on the structural context of the respective subsequences, which are
- exterior - not enclosed by any base pair
- hairpin loop - directly enclosed by a base pair
- non-hairpin loop - subsequence enclosed by two loops forming a bulge, interior or multi-loop
The following figure shows an RNA structure depiction with context annotations (abbreviated by resp. first letter) of unpaired regions that can form RNA-RNA interactions.

IntaRNA can predict single-site interactions within any structural context of the respective subsequences.
| context | exterior | hairpin loop | non-hairpin loop |
|---|---|---|---|
| exterior | |||
| hairpin | |||
| non-hairpin loop |
Note, concatenation-based approaches as implemented in UNAfold, NUPACK or RNAcofold can only predict exterior-exterior context combinations (shown by (b) in the figure above) and are thus not capable to investigate e.g. common loop-exterior or kissing-hairpin-loop interaction patterns that are depicted by (c) and (d) in the figure from above, respectively!
A detailed discussion about different prediction approaches and predictable interaction pattern is available in our publications
- Interactive implementations of thermodynamics-based RNA structure and RNA-RNA interaction prediction approaches for example-driven teaching. and
- Structure and interaction prediction in prokaryotic RNA biology.
Single-site, loop-based RNA-RNA interaction with minimal free energy
This default model of IntaRNA (--model=X) predicts the single-site interaction I with
minimal free energy. That is, it minimizes
arg min ( E_hybrid(I) + ED1(I) + ED2(I) )
I
where E_hybrid represents all energy terms of intermolecular base pairs and
ED corresponds to the energy needed to make the respective subsequences
accessible for inter-molecular base pairing, i.e. removing any possible intra-molecular
base pairs.
The model considers inter-molecular base pair patterns (so called loops) that correspond to (helical) stackings, bulges or interior loops, which are depicted in figure (b) from above. Since intra-molecular base pairs are not explicitely represented, any structural context of single-site interactions is considered/possible within IntaRNA predictions.
This model is used e.g. by the IntaRNA and IntaRNAexact personalities.
Note, the underlying computational model implements a seed-extension strategy
(--model=X, version 3.0+), which first identifies putative seeds that are
subsequently extended to identify the optimal interactions.
Older versions of IntaRNA (version 1.* and 2.*) used a slower optimization
strategy (accessible via --model=S).
Single-site, ensemble-based RNA-RNA interaction with minimal free ensemble energy
IntaRNA supports using --model=P an ensemble based interaction prediction that
is based on partition function computation. To this end, the model computes among all
possible interaction sites S, defined by two interacting subsequences while the ends of the subsequences
form a base pair, the one with maximal partition function Z(S), i.e.
arg max Z(S) = ( exp(-ED1(S)/RT) * exp(-ED2(S)/RT) * sum ( exp(-E_hybrid(I)/RT) ) )
S I of S
which sums the Boltzmann weights of all interactions I (hybridization terms only)
for the given site S multiplied with the Boltzmann weights of the respective
accessibility penalties ED1 and ED2.
Here, R denotes the gas constant and T the temperature of the system.
At T=37 degree Celsius, the product RT is about RT=0.6163173043012.
The site’s partition function can be
used to compute the ensemble energy of all interactions of a given site via
E(S) = -RT log(Z(S)) (log = natural logarithm), which is reported for the optimal site
(see CSV output).
This abstracts from individual inter-molecular base pairing and incorporates the
dynamics and flexibility of the interactions formed by two regions.
The overall partition function is given by
Zall = sum Z(S), i.e. summation over all possible sites,
and enables the computation of the probabilities P_E(I) = exp(-E(I)/RT)/Zall
that a certain interaction I is formed.
Both can be accessed via CSV output.
Note, all computations are only based on the considered interactions that are conform to the currently applied constraints (eg. on seed, accessibility, interaction width, etc.) and do not take all possible interactions into account! Thus, if you are predicting interactions for a subregion only, the results are based on the respective subset of interactions!
This model is used by the IntaRNAens personality.
Helix-based single-site RNA-RNA interaction with minimal free energy
The formation of multiple base pair stackings, i.e. helix formation, requires a ‘winding’ of the respective subsequences. Depending on the structural context, such winding might be sterically and kinetically hindered by the necessary unwinding of intra-molecular structural elements.
The helix-based prediction model aims to incorporate such effects into the predictions of IntaRNA. This is done by restricting the maximum length of inter-molecular helices to a specified number of (stacked) base pairs. That way, ‘wound up’ subhelices are interspaced by flexible interior loops that will allow for a more flexible 3D arrangement of the overall helix.
The following figure depicts the effect of the maximum helix length constraints that only allows for helices up to a specified length. That way, long interactions (left) are avoided and replaced by a more flexible model composed of short inter-molecular helices (right). The blue boxes represent the length-bound helices and the red boxes depict the interspacing unpaired regions (interior loops).

Note, IntaRNA’s helix-block-based model --model=B implements a heuristic, which
only considers for each intermolecular base pair the most stable helix (block) extending
to the right. This results in reduced runtimes while keeping or even improving
the prediction quality in genome wide screens. IntaRNA offers various
helix constraints to guide which helices are considered for interaction
prediction.
This model is used by the IntaRNAhelix personality.
For further details on the model and the underlying algorithm, please refer to our respective publication
Prediction modes
For the prediction of minimum free energy interactions, the following modes
and according features are supported and can be set via the --mode parameter.
The time and space complexities are given for the prediction of two sequences
of equal length n.
| Features | Heuristic --mode=H |
Exact --mode=M |
Seed-only --mode=S |
|---|---|---|---|
| Time complexity (prediction only) | O(n^2) | O(n^4) | O(n^2) |
| Space complexity | O(n^2) | O(n^2) | O(n^2) |
| Seed constraint | |||
| Explicit seeds | |||
| SHAPE reactivity constraint | |||
| No seed constraint | |||
| Minimum free energy interaction | not guaranteed | ||
| Overlapping suboptimal interactions | |||
| Non-overlapping suboptimal interactions |
Note, due to the low run-time requirement of the heuristic prediction mode
(--mode=H), heuristic IntaRNA interaction predictions are widely used to screen
for interaction in a genome-wide scale. If you are more interested in specific
details of an interaction site or of two relatively short RNA molecules, you
should investigate the exact prediction mode (--mode=M) providing the global
minimum free energy interaction.
Putative seed interactions (used by the H and M mode) can be enumerated
and studied using the S mode.
Emulating other RNA-RNA interaction prediction tools
Given these features, we can emulate and extend a couple of RNA-RNA interaction tools using IntaRNA.
TargetScan, RNAhybrid and RNAduplex are approaches that predict the interaction hybrid with minimal interaction energy without consideration whether or not the interacting subsequences are probably involved involved in intramolecular base pairings. Furthermore, no seed constraint is taken into account. This prediction result can be emulated (depending on the used prediction mode) by running IntaRNA when disabling both the seed constraint as well as the accessibility integration using
# prediction results similar to TargetScan/RNAhybrid
IntaRNA [..] --noSeed --acc=N
We add seed-constraint support to TargetScan/RNAhybrid-like computations by removing the
--noSeed flag from the above call.
See IntaRNAexact personality for additional information.
RNAup was one of the first RNA-RNA interaction prediction approaches that took the accessibility of the interacting subsequences into account while not considering the seed feature. IntaRNA’s exact prediction mode is eventually an alternative implementation when disabling seed constraint incorporation. Furthermore, the unpaired probabilities used by RNAup to score the accessibility of subregions are covering the respective overall structural ensemble for each interacting RNA, such that we have to disable accessibility computation based on local folding (RNAplfold). Finally, RNAup only predicts interactions for subsequences of length 25. All this can be setup using
# prediction results similar to 'RNAup -b' (incorporating accessibility of both RNAs)
IntaRNA --mode=M --noSeed --accW=0 --accL=0 --intLenMax=25
We add seed-constraint support to RNAup-like computations by removing the
--noSeed flag from the above call.
See IntaRNAexact personality for additional information.
Limiting memory consumption - window-based prediction
The memory requirement of IntaRNA grows quadratically with lengths of the input sequences. Thus, for very long input RNAs, the requested memory can exceed the available RAM of smaller computers.
This can be circumvented by using a window-based prediction where the input sequences are decomposed in overlapping subsequences (windows) that are processed individually. That way, the maximal memory consumption is defined by the (shorter) window length rather the length of the input sequence, resulting in a user guided memory/RAM consumption.
The window-based computation is enabled by setting the following parameters
--windowWidth: length of the windows/subsequences (value of 0 disables window-based computations)--windowOverlap: overlap of the windows, which has to be larger than the maximal interaction length (see--q|tIntLenMax)
Note, window-based computation produces a computational overhead due to redundant consideration of the overlapping subsequences. Thus, the runtime is increased proportionally to the ratio of window overlap and length.
If only one query and target are given, window-based computation can be parallelized, which typically remedies the computational overhead.
IntaRNA’s multiple personalities
IntaRNA comes as a powerful tool for RNA-RNA interaction prediction supporting various interaction models, prediction modi and constraints. Thus, it is hard for a non-expert user to find and setup the best parameter set for the task at hand.
To provide a better guide how to use IntaRNA, we provide different preset parameter combinations, where each defines one of IntaRNA’s multiple personalities. In the following, we list available personalities and respective use cases.
Each personality can be enabled by either replacing IntaRNA with the respective
personality when calling or by setting the input parameter --personality=....
Thus, depending on how you call IntaRNA, it will react differently.. :smile:
IntaRNA
The standard personality of IntaRNA implements the prediction approach of (Busch et al., 2008) with the extension from (Mann et al., 2017). Thus it uses a fast heuristic prediction mode in combination with local accessibility estimates. It predicts minimum free energy RNA-RNA interactions for single-site unconstraint interaction models.
Thus, IntaRNA provides (typically) optimal predictions on a single inter-molecular base pair level. Output is per default restricted to the interaction site and base pairs, since intra-molecular structure is implicitely considered via the accessibility penalties represented by the ED terms of the energy calculation (see detailed output).
IntaRNA1 and IntaRNA2
For backward compatibility, we also provide the personality IntaRNA1 and IntaRNA2,
which emulate the settings of the respective version with the currently
available parameters.
Both personalities make use of the slower --model=S
prediction strategy first
introduced for IntaRNA version 1.0.
Note, IntaRNA v1.* used the old Turner99 energy parameters, such that the
--energyVRNA=Turner99 energy set is preset, when using IntaRNA1.
IntaRNAexact
RNAup was the first accessibility-based RNA-RNA interaction prediction approach and it implements methods to identify the optimal minimum free energy interaction for unconstraint, single-site interaction models. IntaRNA also implements analogous methods when exact prediction modes are used, and thus can provide RNAup-like predictions.
IntaRNAexact therefore
- uses exact prediction mode,
- restricts the maximal interaction length to 60,
- uses global accessibility values (reenable local computation respectively if needed, e.g. for mRNAs or genomic subsequences) and
- reports suboptimal interactions that can overlap in both RNAs.
In contrast to RNAup, it also
- enforces seed constraints,
- uses a much faster seed-extension-based computation model,
- allows longer interaction length, and
- enables much more flexible output options.
If needed, you can disables these features.
Thus, IntaRNAexact produces RNAup-like predictions with the extensive output and constraint options of IntaRNA. Since exact computations are computationally much more demanding, IntaRNAexact is slower than IntaRNA. Therefore, you should use IntaRNAexact if you want to investigate the details of a single or few interactions unbiased by the heuristics applied in normal IntaRNA. If you are using long RNAs, you should constraint predictions to the regions of interest or constraint the seed regions.
In summary, the following calls are equivalent.
IntaRNAup ...
IntaRNA --personality=IntaRNAup ...
IntaRNA --model=X --mode=M --accW=0 --accL=0 --intLenMax=60 --outOverlap=B ...
IntaRNAhelix
IntaRNAhelix provides helix-based RNA-RNA interaction prediction described in (Gelhausen et al., 2019). It therefore enables per default
All other parameters are kept from the normal IntaRNA personality, such that IntaRNAhelix predicts per default interactions heuristically based on optimal helix blocks, which is faster than the normal mode but applies more constraints. Thus, you should use IntaRNAhelix if you want to focus predictions on stable subinteractions (helices) and need to do it fast.
In summary, the following calls are equivalent.
IntaRNAhelix ...
IntaRNA --personality=IntaRNAhelix ...
IntaRNA --model=B ...
IntaRNAduplex
TargetScan, RNAhybrid and RNAduplex are approaches that predict the RNA-RNA interaction with minimal free energy without consideration whether or not the interacting subsequences are accessible. Furthermore, no seed constraint is taken into account.
IntaRNAduplex provides analogous predictions by
- disabling accessibility consideration.
In constrast to other approaches, it
- uses a fast heuristic prediction mode, and
- enforces seed constraints.
Thus, IntaRNAduplex is useful if you are not interested in the structuredness of the interacting molecules, either since they are very short or very long. The latter makes accessibility prediction difficult, since it is not only governed by thermodynamics.
In summary, the following calls are equivalent.
IntaRNAduplex ...
IntaRNA --personality=IntaRNAduplex ...
IntaRNA --qAcc=N --tAcc=N ...
IntaRNAsTar
IntaRNAsTar provides optimized parameters for large scale (genome-wide) sRNA target prediction identified via the benchmark introduced in our publication (Raden et al., 2020). This covers
- no GU base pairs in seeds
- minimal unpaired probability of 0.001 of seed regions
- maximal interaction length of 60
- maximal interior loop size of 8
- minimal unpaired probability of 0.001 of interacting regions
Furthermore, it ensures
In summary, the following calls are equivalent.
IntaRNAsTar ...
IntaRNA --personality=IntaRNAsTar ...
IntaRNA --intLenMax=60 --intLoopMax=8 --seedNoGU --seedMinPu=0.001 --outMinPu=0.001 --outNoLP --outNoGUend --outOverlap=Q --outMode=C --outCsvCols=id1,id2,start1,end1,start2,end2,E ...
IntaRNAseed
IntaRNAseed only considers and reports putative seed interactions. To this end, it
In summary, the following calls are equivalent.
IntaRNAseed ...
IntaRNA --personality=IntaRNAseed ...
IntaRNA --mode=S ...
IntaRNAens
IntaRNAens provides ensemble-based predictions, i.e. identifies an optimal site rather than an individual optimal interaction. This abstracts from individual base pairing and incorporates the base pairing flexibility of interacting regions. To do so, it
In summary, the following calls are equivalent.
IntaRNAens ...
IntaRNA --personality=IntaRNAens ...
IntaRNA --model=P ...
How to constrain predicted interactions
Interaction restrictions
The predicted RNA-RNA interactions can be enhanced if additional knowledge is available. To this end, IntaRNA provides different options to restrict predicted interactions.
One of the most general restriction is the maximal energy (inversely related to
stability) an RNA-RNA interaction is allowed to have. Per default, a reported
interaction should have a negative energy (<0) to be energetically favorable.
This report barrier can be altered using --outMaxE. For suboptimal interaction
restriction, please refer to suboptimal interaction prediction section.
Another stability constraint is --outNoLP, which forbids lonely, i.e. non-stacked,
inter-molecular base pairs. These are typically not contributing much to the
overall stability and can lead to instable subinteractions when e.g. enclosed
by two large interior loops.
In addition, with --outNoGUend one can prohibit weak GU base pairs at
interaction ends and within interior loops.
That way, only stable inter-molecular helix ends are considered.
If you are only interested in predictions for highly accessible regions, i.e.
with a high probability to be unpaired, you can use the --outMinPu parameter.
If given, each individual position of the interacting subsequences has to have
an unpaired probability reaching at least the given value. This significantly
increases prediction time but will exclude predictions where the formation of
the interaction (intermolecular base pairing) replaces intramolecular base
pairing (where the latter will cause low unpaired probabilities for the
respective positions).
Furthermore, the region where interactions are supposed to occur can be restricted
for target and query independently. To this end, a list of according
subregion-defining index pairs
can be provided using --qRegion and --tRegion, respectively. The indexing
starts with 1 (or the value set via --qIdxPos0 and --tIdxPos0)
and should be in the format from1-end1,from2-end2,.. using
integers.
Note, if you want to have predictions individually for each region
combination (rather than just the best for each query-target combination) you
want to add --outPerRegion to the call.
If you are dealing with very long sequences it might be useful to use the
automatic identification of accessible regions, which dramatically reduces
runtime and memory consumption of IntaRNA since predictions are only done for
individual regions and not for the whole sequence. Here, we use a
heuristic approach that finds and ignores subregions that are unlikely to form
an interaction, resulting in a decomposition of the full sequence range into
intervals of accessible regions. It can be enabled by providing the maximal
length of the resulting intervals via the parameters --qRegionLenMax and
--tRegionLenMax.
More specifically, starting from the full
sequence’s index range, the algorithm iteratively identifies in every too-long
range the window with highest ED value (penalty for non-accessibility). To
this end, it uses windows of length --seedBP to find subsequences where it is
most unlikely that a seed might be formed. This window is removed from the range,
which results in two shorter ranges. If a range is shorter than --seedBP, it
is completely removed.
Finally, it is possible to restrict the overall length an interaction is allowed
to have via --intLenMax. This can be done independently for the query and target sequence using
--qIntLenMax and --tIntLenMax, respectively. By setting to 0 (default),
the smaller of the full sequence length and the maximal accessibility-window
size is used (see --accW, --tAccW, or --qAccW).
Seed constraints
For different types of RNA-RNA interactions it was found that experimentally verified interactions were showing a short and compact subinteraction of high stability (= low energy). It was hypothesized that these regions are among the first formed parts of the full RNA-RNA interaction, and thus considered as the seed of the overall interaction.
Based on this observation, RNA-RNA interaction predictors were enhanced by incorporating such seed constraints into their prediction pipeline, i.e. a reported interaction has to feature at least one seed. Typically, a seed is defined as a short subinteraction of 5-8 consecutive base pairs that are not enclosing any unpaired nucleotides (or if so only very few).
IntaRNA supports the definition of such seed constraints and adds further options to even more constrain the seed selection. The list of options is given by
--seedBP: the number of base pairs within the seed--seedMaxUP: the maximal overall number of unpaired bases within the seed--seedQMaxUP: the maximal number of unpaired bases within the query’s seed region--seedTMaxUP: the maximal number of unpaired bases within the target’s seed region--seedMaxE: the maximal overall energy of the seed (to exclude unlikely seed interactions)--seedMaxEhybrid: the maximal hybridization energy (including E_init) of the seed (to exclude weak seed hybridizations likely to fall off again)--seedMinPu: the minimal unpaired probability of each seed region in query and target--seedQRange: a list of index intervals where a seed in the query is allowed--seedTRange: a list of index intervals where a seed in the target is allowed--seedNoGU: if present, no GU base pairs are allowed within seeds--seedNoGUend: if present, no GU base pairs are allowed at seed ends
Alternatively, you can set
--seedTQ: to specify explicit seed interactions
Seed constraint usage can be globally disabled using the --noSeed flag.
Explicit seed input
Some experiments provide hints or explicit knowledge about the seed or
even provide details about some intermolecular base pairs formed between two RNAs.
This information can be incorporated into IntaRNA predictions by providing
explicit seed information. To this end, the --seedTQ parameter can be used.
It takes a comma-separated list of seed string encodings in the format
startTbpsT&startQbpsQ, which is in the same format as the IntaRNA hybridDB
output (see below), i.e. e.g. --seedTQ='4|||.|&7||.||'
(ensure you quote the seed encoding to avoid a shell interpretation of the pipe symbol ‘|’)
to encode a seed interaction like
the following
target
4 8
| |
5'-AAAC C UGGUUUGG-3'
AC C C
|| | |
UG G G
3'-GGUU U CCCACAAA-5'
| |
11 7
query
If several or alternative seeds are known, you can provide all as a comma-separated list and IntaRNA will consider all interactions that cover at least one of them.
Helix constraints
For helix-based interaction models, IntaRNA provides various constraints for the helices considered for interaction prediction:
-
--helixMinBP: minimal number of base pairs inside a helix (mindefault=2) --helixMaxBP: maximal number of base pairs inside a helix--helixMaxE: maximal energy of each helix; any helix with higher energy is not considered for interaction prediction. Note, this constraint is useful to restrict the prediction to more stable interactions. See also--helixFullE.--helixFullEif present, the overall helix energy (including E_init, ED, dangling end contributions, etc.) is used for energy checks (--helixMaxE). Otherwise, only the loop-terms are considered.--helixMinPu: minimal unpaired probability (per helix subsequence); thus, one can constrain prediction to helices in accessible regions.--helixMaxIL: maximal size for each internal loop size in a helix; that way you can relax the helix definition and allow for a given number of unpaired bases between consecutive base pairs within a helix. Note, increasing this value reduces the impact of the helix-based model for prediction.
SHAPE reactivity data to enhance accessibility computation
For some RNA sequences, experimental reactivity data is available that can be
used to guide/help the structure and thus accessibility prediction for the RNA
molecule. IntaRNA supports such data by interfacing the Vienna RNA package
capabilities for SHAPE reactivity data incorporation, see
Lorenz et al. (2015, 2016) or the
RNAfold manpage.
To use this feature, ViennaRNA-based accessibility computation is needed,
i.e. --t|qAcc=C, which is the default mode (no action required).
The SHAPE reactivity data can be provided via file using --qShape or
--tShape for query or target sequence, respectively. The input format is a
space-separated 3-column format providing position, nucleotide and reactivity value as
exmemplified below.
1 G 0.134
2 C 0.044
3 C 0.057
4 G 0.114
5 U 0.094
6 C 0.035
7 G 0.909
8 C 0.224
9 C 0.529
10 A 1.475
Independently for each sequence, it is possible
to define the methods to be used to convert the data into pseudo energies and
pairing probabilities. The respective IntaRNA arguments are
--qShapeMethod|--tShapeMethod
and --qShapeConversion|--tShapeConversion, which mimics the according
tool arguments in the Vienna RNA package (see e.g. the
RNAfold manpage).
An example call that shows the effect of SHAPE data incorporation is given below (assuming the SHAPE data from above is stored in
the file data-SHAPE.txt).
IntaRNA -q GCCGUCGCCA -t GCCGUCGCCA --tShape=data-SHAPE.txt --out=qAcc:STDOUT --out=tAcc:STDERR --out=/dev/null
For further details, please refer to our respective publication
Output setup
Output modes
The RNA-RNA interactions predicted by IntaRNA can be provided in different
formats. The style is set via the argument --outMode and the different modes
will be discussed below.
Furthermore, it is possible to define where to output, i.e. using --out
you can either name a file or one of the stream names STDOUT|STDERR. Note,
any string not matching one of the two stream names is considered a file name.
The file will be overwritten by IntaRNA!
If a file name ends in .gz, gzip-compressed binary output is generated.
Besides interaction output, you can set the verbosity of computation information
using the -v or --verbose arguments. To reduce the output to a minimum, you
can redirect all logging output of user information, warnings or verbose output
to a specific file using --default-log-file=LOGFILENAME (no gzip compression
supported).
If you are not interested in any logging output, redirect it to nirvana via
--default-log-file=/dev/null. Note, error output is not redirected and always
given on standard output streams.
Standard RNA-RNA interaction output with ASCII chart
The standard output mode --outMode=N provides a detailed ASCII chart of the
interaction together with its overall interaction energy.
For an example see the Just run … section.
Detailed RNA-RNA interaction output with ASCII chart
Using --outMode=D, a detailed ASCII chart of the interaction together with
various interaction details will be provided. An example is given below.
# call: IntaRNA -t AAACACCCCCGGUGGUUUGG -q AAACACCCCCGGUGGUUUGG --outMode=D --seedBP=4
target
5 16
| |
5'-AAAC C U UUGG-3'
ACC CCGG GGU
||| ++++ |||
UGG GGCC CCA
3'-GGUU U C CAAA-5'
| |
16 5
query
interaction seq1 = 5--16
interaction seq2 = 5--16
interaction energy = -6.76 kcal/mol
= E(init) = 4.1
+ E(loops) = -18.8
+ E(dangleLeft) = -0.6
+ E(dangleRight) = -0.6
+ E(endLeft) = 0.5
+ E(endRight) = 0.5
: E(hybrid) = -14.9
+ ED(seq1) = 4.07
: Pu(seq1) = 0.00135534
+ ED(seq2) = 4.07
: Pu(seq2) = 0.00135534
seed seq1 = 9--12
seed seq2 = 9--12
seed energy = -1.42
seed ED1 = 2.66
seed ED2 = 2.66
seed Pu1 = 0.0133541
seed Pu2 = 0.0133541
Note, within this output seq1 and respective values refer to the target
sequence and seq2 etc. to the query RNA.
Position annotations start indexing with 1 at the 5’-end of each RNA
(or the respective value set via --qIdxPos0 and --tIdxPos0).
ED values are the energy penalties for reduced accessibility
and Pu denote unpaired probabilities of the respective interacting subsequences.
Note, Pu values are recomputed from rounded ED values and are thus not equal
to the RNAplfold values from which the ED values are derived from!
Base pairs that are part of putative seed interactions are represented by
+ symbols within the interaction chart. Note, an interaction can contain more
than one putative seed and all respective base pairs are highlighted.
In addition, the seed information (given at the end of the output) will provide
information for all possible seeds sorted by increasing energy (ie. the most
stable and thus most likely seed is always given left most).
If you are only interested in the most stable seed, you should add
--outBestSeedOnly to the call.
Customizable CSV RNA-RNA interaction output
IntaRNA provides via --outMode=C a flexible interface to generate RNA-RNA
interaction output in CSV format (using ; as separator). Note, target sequence
information is listed with index 1 while query sequence information is given
by index 2.
# call: /IntaRNA -t AAACACCCCCGGUGGUUUGG -q AAACACCCCCGGUGGUUUGG --outMode=C --seedBP=4 --outOverlap=B -n 3
id1;start1;end1;id2;start2;end2;subseqDP;hybridDP;E
target;5;16;query;5;16;ACCCCCGGUGGU&ACCCCCGGUGGU;(((.((((.(((&))).)))).)));-6.76
target;6;16;query;5;15;CCCCCGGUGGU&ACCCCCGGUGG;((.((((.(((&))).)))).));-5.56
target;7;16;query;5;15;CCCCGGUGGU&ACCCCCGGUGG;((((((.(((&))).)))).));-5.55
For each prediction, a row in the CSV is generated. The column separator within
the tabular CSV output can be changed using --outSep, e.g. to produce tab-separated
.tsv output.
Using the argument --outCsvCols, the user can specify what columns are
printed to the output using a comma-separated list of colIds. Available colIds
are
id1: id of first sequence (target)id2: id of second sequence (query)seq1: full first sequenceseq2: full second sequencesubseq1: interacting subsequence of first sequencesubseq2: interacting subsequence of second sequencesubseqDP: hybrid subsequences compatible with hybridDPsubseqDB: hybrid subsequences compatible with hybridDBstart1: start index of hybrid in seq1end1: end index of hybrid in seq1start2: start index of hybrid in seq2end2: end index of hybrid in seq2hybridDP: hybrid in VRNA dot-bracket notation (interaction sites only)hybridDPfull: hybrid in VRNA dot-bracket notation (full sequence length)hybridDB: hybrid in dot-bar notation (interactin sites only)hybridDBfull: hybrid in dot-bar notation (full sequence length)bpList: list of hybrid base pairs, e.g. ‘(4,3):(5,2):(7,1)’E: overall interaction energy = E_hybrid + ED1 + ED2ED1: ED value of seq1ED2: ED value of seq2Pu1: probability to be accessible for seq1Pu2: probability to be accessible for seq2E_init: initiation energyE_loops: sum of loop energies (excluding E_init)E_dangleL: dangling end contribution of base pair (start1,end2)E_dangleR: dangling end contribution of base pair (end1,start2)E_endL: penalty of closing base pair (start1,end2)E_endR: penalty of closing base pair (end1,start2)E_hybrid: energy of hybridization only = E - ED1 - ED2E_norm: length normalized energy = E / ln(length(seq1)*length(seq2))E_hybridNorm: length normalized energy of hybridization only = E_hybrid / ln(length(seq1)*length(seq2))E_add: user defined energy correction term incorporated intoEw: Boltzmann weight ofE, e.g. used for partition function computationseedStart1: start index of the seed in seq1 (* see below)seedEnd1: end index of the seed in seq1 (* see below)seedStart2: start index of the seed in seq2 (* see below)seedEnd2: end index of the seed in seq2 (* see below)seedE: overall energy of the seed only (including seedED etc) (* see below)seedED1: ED value of seq1 of the seed only (excluding rest) (* see below)seedED2: ED value of seq2 of the seed only (excluding rest) (* see below)seedPu1: probability of seed region to be accessible for seq1 (* see below)seedPu2: probability of seed region to be accessible for seq2 (* see below)Eall: ensemble energy of all considered interactions (-RT*log(Zall))Zall: partition function of all considered interactionsEall1: ensemble energy of all considered intra-molecular structures of seq1 (given its accessibility constraints)Eall2: ensemble energy of all considered intra-molecular structures of seq2 (given its accessibility constraints)EallTotal: total ensemble energy of all considered interactions including the ensemble energies of intra-molecular structure formation (Eall+Eall1+Eall2)Etotal: total energy of an interaction including the ensemble energies of intra-molecular structure formation (E+Eall1+Eall2)Zall1: partition function represented byEall1(exp(-Eall1/RT))Zall2: partition function represented byEall2(exp(-Eall2/RT))P_E: probability of an interaction (site) within the considered ensembleRT: normalized temperature used for Boltzmann weight computation
(*) Note, since an interaction can cover more than one seed, all seed* columns
might contain multiple entries separated by ‘:’ symbols. In order to print only
the most stable (lowest seed energy) seed information add --outBestSeedOnly
to the call.
Note further, Pu values are recomputed from rounded ED values and are thus not equal
to the RNAplfold values from which the ED values are derived from!
Using --outCsvCols '*', all available columns are added to the output.
Energies are provided in unit kcal/mol, probabilities in the interval [0,1].
Position annotations start indexing with 1 (or the values set via --qIdxPos0 and --tIdxPos0).
The hybridDP format is a dot-bracket notation as e.g. generated by RNAup.
Here, for each target sequence position within the interaction,
a ‘.’ represents a position not involved
in the interaction while a ‘(‘ marks an interacting position. For the query
sequence this is done analogously but using a ‘)’ for interacting positions.
Both resulting strings are concatenated by a separator ‘&’ to yield a single
string encoding of the interaction’s base pairing details.
The hybridDB format is similar to the hybridDP but also provides site information.
Here, a bar ‘|’ is used in both base pairing encodings (which makes it a ‘dot-bar encoding’).
Furthermore, each interaction string is prefixed
with the start position of the respective interaction site.
In the following, an altered CSV output for the example from above is generated.
# call: IntaRNA --outCsvCols=Pu1,Pu2,subseqDB,hybridDB -t AAACACCCCCGGUGGUUUGG -q AAACACCCCCGGUGGUUUGG --outMode=C --seedBP=4 --outOverlap=B -n 3
Pu1;Pu2;subseqDB;hybridDB
0.00135534;0.00135534;5ACCCCCGGUGGU&5ACCCCCGGUGGU;5|||.||||.|||&5|||.||||.|||
0.00135534;0.00135534;6CCCCCGGUGGU&5ACCCCCGGUGG;6||.||||.|||&5|||.||||.||
0.00137751;0.00135534;7CCCCGGUGGU&5ACCCCCGGUGG;7||||||.|||&5|||.||||.||
You can produce a sorted CSV output using the argument --outCsvSort=..,
which provides the colId from the CSV output to be used for sorting.
Below, you find the hybridDB-sorted output from above.
# call: IntaRNA --outCsvCols=Pu1,Pu2,subseqDB,hybridDB --outCsvSort=hybridDB -t AAACACCCCCGGUGGUUUGG -q AAACACCCCCGGUGGUUUGG --outMode=C --seedBP=4 --outOverlap=B -n 3
Pu1;Pu2;subseqDB;hybridDB
0.00135534;0.00135534;5ACCCCCGGUGGU&5ACCCCCGGUGGU;5|||.||||.|||&5|||.||||.|||
0.00135534;0.00135534;6CCCCCGGUGGU&5ACCCCCGGUGG;6||.||||.|||&5|||.||||.||
0.00137751;0.00135534;7CCCCGGUGGU&5ACCCCCGGUGG;7||||||.|||&5|||.||||.||
Ensemble output of considered RNA-RNA interactions
The output mode --outMode=E provides information on the ensemble of all
considered RNA-RNA interactions (e.g. compatible with the current seed constraints).
This covers key-value pairs of the following information using
column labels introduced for the CVS output:
id1: id of first sequence (target)id2: id of second sequence (query)RT: the scaled temperature used for Boltzmann-weight computationEall: ensemble energy of all considered interactions (-RT*log(Zall))Eall1: ensemble energy of all considered intra-molecular structures of seq1 (given its accessibility constraints)Eall2: ensemble energy of all considered intra-molecular structures of seq2 (given its accessibility constraints)EallTotal: total ensemble energy of all considered interactions including the ensemble energies of intra-molecular structure formation (Eall+Eall1+Eall2)
Note, Zall depends on the selected
prediction mode and
RNA-RNA interaction model.
It holds
Zall(--model=S) <= Zall(--model=P)as well asZall(--mode=H --model=P) <= Zall(--mode=M --model=P).
Thus, most accurate results are computed using
IntaRNA --model=P --mode=M --outMode=E ...
BUT: the seed-extension strategy in --model=X does not allow for correct Zall computation. Thus, avoid Eall or Zall output in this mode!
Pairwise vs. all-vs-all
When multiple query and target sequences are provided, IntaRNA predicts interactions for all pairs of query-target combinations, i.e. all-vs-all.
Alternatively, you can enforce pairwise processing using --outPairwise.
When given, sequences are combined based on their input order, i.e. the 5th target is
(only) considered for interaction prediction with the 5th query sequence. Thus, you
have to provide the same number of query and target sequences.
Sequence indexing
Per default, IntaRNA assumes sequence indexing to be natural, i.e. starting with index 1 at the RNAs’ 5’-ends.
If needed, you can alter indexing (independently for query and target) using the
--qIdxPos0 and --tIdxPos0 parameters, respectively. That way you can
- start indexing at an arbitrary position, e.g. for subsequences of long RNAs or genomes,
- get a 0-based indexing if needed by your down-stream processing, or
- get relative indexing, e.g. relative to the start codon within an mRNA sequence.
The latter, i.e. relative sequencing, starts increasing indexing with the given first (negative) index but omits the ‘0’ index. Thus, position ‘-1’ is followed by ‘+1’. An example for a start-codon-related indexing is given below, where the start codon within the target is located at position 9.
# call: IntaRNA -t gacugguaAUGggac --tIdxPos0=-8 -q UCUUACCGUGAGUC --seedBP=5
target
-8 1
| |
5'- --- UGGGAC-3'
GACU GGUAA
:||| +++++
CUGA CCAUU
3'- GUG CU-5'
| |
14 3
query
interaction energy = -6.39 kcal/mol
Suboptimal RNA-RNA interaction prediction and output restrictions
Besides the identification of the optimal (e.g. minimum-free-energy) RNA-RNA
interaction, IntaRNA enables the enumeration of suboptimal interactions. To this
end, the argument -n N or --outNumber=N can be used to generate up to N
interactions for each query-target pair (including the optimal one).
Note: suboptimal interaction enumeration is not exhaustive! That is, for each interaction site (defined by the left- and right-most intermolecular base pair) only the best interaction is reported! In heuristic prediction mode (default mode of IntaRNA), this is even less exhaustive, since only for each left-most interaction boundary one interaction is reported!
Furthermore, it is possible to restrict (sub)optimal enumeration using
--outMaxE: maximal energy for any interaction reported--outDeltaE: maximal energy difference of suboptimal interactions’ energy to the minimum free energy interaction--outOverlap: defines if and where overlapping of reported interaction sites is allowed:- ‘N’ : no overlap neither in target nor query allowed for reported interactions
- ‘B’ : overlap allowed for interacting subsequences for both target and query
- ‘T’ : overlap allowed for interacting subsequences in target only
- ‘Q’ : overlap allowed for interacting subsequences in query only
Note: non-overlapping output (i) is heuristic by considering for each left interaction site only the best right extension for overlap computation and (ii) increases runtime. To get optimized results of non-overlapping suboptimals, rerun IntaRNA and mark the optimal (mfe) interaction region as blocked.
Energy parameters and temperatures
The selection of the correct temperature and energy parameters is cruicial for a correct RNA-RNA interaction prediction. To this end, various settings are supported by IntaRNA.
The temperature can be set via --temperature=Cto set a temperature C in
degree Celsius. Note, this is important especially for predictions within plants
etc., since the default temperature is 37C.
The energy model used can be specified using the --energy parameters using
- ‘B’ for base pair maximization similar to the Nussinov intramolecular structure prediction.
Here, each base pair contributes an energy term of
-1independently of its structural or sequence context. This mode is mainly useful for study or teaching purpose. - ‘V’ enables Nearest Neighbor Model energy computation similar to the Zuker
intramolecular structure prediction using the Vienna RNA package routines.
Within this model, the energy contribution of a base
pair depends on its directly enclosed (neighbored) basepair and the subsequence(s)
involved. Different energy parameter sets have been experimentally derived
in the last decades. Since IntaRNA makes use of the energy evaluation routines
of the Vienna RNA package, all parameter sets from the Vienna RNA package are
available for RNA-RNA interaction prediction. Per default, the default parameter
set of the linked Vienna RNA package version is used. You can change the parameter
set using the
--energyVRNAparameter as explained below.
If Vienna RNA package is used for energy computation (--energy=V), per default
the default parameter set of the linked Vienna RNA package is used (e.g. the
Turner04 set for VRNA 3.0.0). If you want to use a different parameter set, you
can provide an according parameter file via --energyVRNA=MyParamFile. The
following example shows how to run an IntaRNA-v1-like prediction (default for the
IntaRNA1 personality) using the old Turner99 parameter set (as used by IntaRNA v1.*).
# IntaRNA v1.* like energy parameter setup
IntaRNA --energyVRNA=Turner99
# alternative IntaRNA v1.* like energy parameter setup with explicit file
IntaRNA --energyVRNA=/usr/local/share/Vienna/rna_turner1999.par
IntaRNA provides the following predefined --energyVRNA values that load respective
parameter sets from the Vienna RNA package without explicit file specification:
Turner04=rna_turner2004.parTurner99=rna_turner1999.parAndronescu07=rna_andronescu2007.parIf no value for--energyVRNAis provided, the default model of the underlying Vienna RNA package is used (see respective documentation).
To increase prediction quality and to reduce the computational complexity, the
number of unpaired bases between intermolecular base pairs is restricted via
--intLoopMax (similar to internal loop length restrictions in single RNA folding algorithm). The
upper bound can be set independently for the query and target sequence via
--qIntLoopMax and --tIntLoopMax, respectively, and defaults to 16.
Note, you can shift the energy spectrum using --energyAdd=... The given value
is always added when the overall energy of an interaction is computed. That way,
you can correct for conditional predictions based on given
accessibility constraints.
If lonely (non-stacked) inter-molecular base pairs are of no interest, since they
can lead to instable subinteraction when enclosed by two large interior loops,
one can disable their prediction using --outNoLP.
For some applications it is needed to ignore dangling end energy contributions
that reflect minor stabilizing (stacking) contributions of unpaired bases
adjacent to a helix-closing base pair.
They can be ignored via --energyNoDangles.
Additional output files
IntaRNA enables the generation of various additional information in dedicated
files/streams. The generation of such output is guided by an according (repeated)
definition of the --out argument in combination with one of the following
argument prefixes (case insensitive) that have to be colon-separated to the
targeted file/stream name:
qSpotProb:/tSpotProb:query/target’s spot probability profile (CSV format), respectivelyspotProb:all spot probabilities (CSV format)qMinE:/tMinE:the query/target’s minimal interaction energy profile (CSV format), respectivelypMinE:minimal interaction energy for all query-target index pairs (CSV format)qAcc:/tAcc:the query/target’s ED accessibility values (RNAplfold-like format), respectivelyqPu:/tPu:the query/target’s unpaired probabilities (RNAplfold format; rounded!!), respectively
Note, for multiple sequences in FASTA input, the provided file names are suffixed by
-t#q#: for*spotProband*minEoutput, and-s#: for*Accand*Puoutput, where#denotes the according target/query sequence number within the input where numbering starts with 1.
The column separator within tabular CSV output (defaulting to ;) can be changed
using --outSep, e.g. to produce tab-separated .tsv output.
Note further, qPu:|tPu: will report unpaired probability values based on rounded accessibility (ED) values.
Thus, these values will most likely differ from values eg. produced by the program RNAplfold.
We therefore strongly recommend to store qAcc:|tAcc: values when you want to use them
as input for subsequent IntaRNA calls!
As for normal output, if the specified file name ends in .gz, gzip-compressed
binary output is generated. This is especially useful for large output data like
accessibility or unpaired probability information as well as pairwise energy
or spot probability profiles.
Minimal energy profiles
To get a more global view of possible interaction sites for a pair of interacting RNAs, one can generate the minimal energy profile for each sequence (independently).
For instance, to generate the target’s profile, add the following to your IntaRNA
call: --out=tMinE:MYPROFILEFILE.csv. For the query’s profile, use --out=qMinE:.. respectively.
This will produce an according CSV-file (; separated) with the according minimal
energy profile data that can be visualized with any program of your liking.
In the following, such an output was visualized using R:
d <- read.table("MYPROFLEFILE.csv", header=T, sep=";");
plot( d[,1], d[,3], xlab="sequence index", ylab="minimal energy", type="l", col="blue", lwd=2)
abline(h=0, col="red", lty=2, lwd=2)
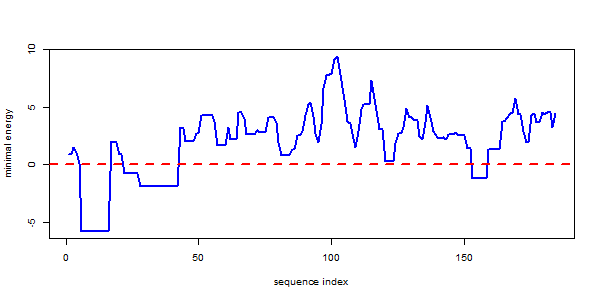
This plot reveals two less but still stable (E below 0) interaction sites beside the mfe interaction close to the 5’-end of the molecule.
Minimal energy for all intermolecular index pairs
To investigate how stable RNA-RNA interactions are distributed for a given pair
of RNAs, you can also generate the minimal energy for all intermolecular index
pairs using --out=pMinE:MYPAIRMINE.csv. This generates a CSV file (;separated)
holding for each index pair the minimal energy of any interaction covering this
index combination or NA if no covers it at all.
This information can be visualized with your preferred program. In the following, the provided R call is used to generate a heatmap visualization of the RNA-RNA interaction possibilities.
# read data, skip first column, and replace NA and E>0 values with 0
d <- read.table("MYPAIRMINE.csv",header=T,sep=";");
d <- d[,2:ncol(d)];
d[is.na(d)] = 0;
d[d>0] = 0;
# plot
image( 1:nrow(d), 1:ncol(d), as.matrix(d), col = heat.colors(100), xlab="index in sequence 1", ylab="index in sequence 2");
box();
The following plot (for the minimal energy profile example from above) reveals, that the alternative stable (E<0) interactions all involve the mfe-site in the second sequence and are thus less likely to occure.
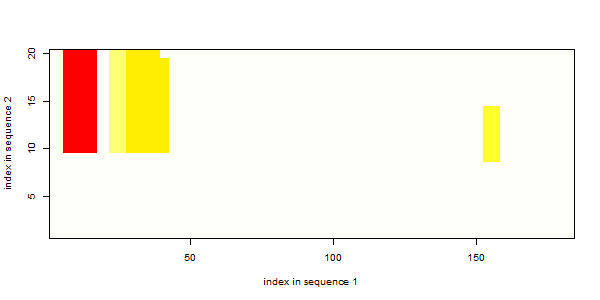
A more sophisticated plot can be done using ggplot2 from the tidyverse package.
library(tidyverse)
read_delim("pairMinE-t1q1.csv", delim=";") %>%
replace(is.na(.), 0) %>%
rename( target = minE ) %>%
pivot_longer( cols=-target, names_to = "query", values_to = "E" ) %>%
separate( query, into = c("qSeq","qIdx"), sep = "_") %>%
separate( target, into = c("tSeq","tIdx"), sep = "_") %>%
mutate(across(c(tIdx,qIdx,E),as.numeric)) %>%
mutate(E = ifelse( E>0, 0, E)) %>%
ggplot(aes(x=tIdx,y=qIdx, fill=E)) +
geom_tile() +
scale_fill_gradient(low="blue",high="red")
Spot probability profiles
Similarly to minimal energy profiles, it is also possible to compute position-wise probabilities how likely a position is covered by an interaction, i.e. its spot probability. To the end, we compute for each position i the partition function Zi of all interactions covering i. Given the overall partition function Z including all possible interactions, the position-speficit spot probability for i is given by Zi/Z.
Such profiles can be generated using --out=qSpotProb:MYPROFILEFILE.csv or
--out=tSpotProb:... for the query/target sequence respectively and independently.
Note, instead of a file you can also write the profile to stream using STDOUT
or STDERR instead of a file name.
Interaction probabilities for interaction spots of interest
For some research questions, putative regions of interactions are known from other sources and it is of interest to study the effect of competitive binding or other scenarios that might influence the accessibility of the interacting RNAs (e.g. refer to SHAPE data or structure/accessibility constraints).
To this end, one can specify the spots of interest by intermolecular index pairs,
e.g. using 5&67 to encode the fifth target RNA position (first number of the
encoding) and the 67th query RNA
position (second number of the encoding). Note, indexing starts with 1
(or the values set via --qIdxPos0 and --tIdxPos0).
Multiple spots can be provided as comma-separated list. The list in
concert with an output stream/file name (colon-separated) can be passed via the
--out argument using the spotProb: prefix, e.g.
IntaRNA ... --out=spotProb:5&67,33&12:mySpotProbFile.csv
The reported probability is the ratio of according partition functions. That is,
for each interaction I that respects all input constraints and has an energy
below 0 (or set --outMaxE value) the respective Boltzmann weight bw(I)
is computed by bw(I) = exp( - E(I) / RT ). This weight is added to the
overallZ partition function. Furthermore, we add the weight to a respective
spot associated partition function spotZ, if the interaction I spans the spot, ie.
the spot’s indices are within the interaction subsequences of I. If none of
the spots if spanned by I, the noSpotZ partition function is increased by
bw(I). The final probability of a spot is than given by spotZ/overallZ and
the probability of interactions not covering any of the tracked spots is
computed by noSpotZ/overallZ and reported for the pseudo-spot encoding 0&0
(if indexing starts with 1) or (--qIdxPos0-1)&(--tIdxPos0-1) if the respective
parameters for index shifting are set.
NOTE and be aware that the probabilities are only estimates since IntaRNA is not considering (in default prediction mode) all possible interactions due to its heuristic (see discussion about suboptimal interactions). Nevertheless, since the Boltzmann probabilities are dominated by the low(est) energy interactions, we consider the probability estimates as meaningful!
Spot probabilities for all intermolecular index pairs
If interested in all intermolecular index pair combinations (all spots), you
can exclude the list from the call and only specify the output file/stream by
--out="spotProb:MYSPOTPROBFILE.csv". The resulting semicolon-separated table provides the spot
probability for each index pair combination, as shown below.
# call : IntaRNA --out="spotProb:STDOUT" -t AAACACCCCCGGUGGUUUGG -q AAACACCCCCGGUGGUUUGG --energy=B -m M --seedBP=3 --out=/dev/null
spotProb;A_1;A_2;A_3;C_4;A_5;C_6;C_7;C_8;C_9;C_10;G_11;G_12;U_13;G_14;G_15;U_16;U_17;U_18;G_19;G_20
A_1;1.14168e-05;3.62052e-05;6.5358e-05;0.000125994;0.000196552;0.000328442;0.000553431;0.000824511;0.00120974;0.00133627;0.00134247;0.00135501;0.00140472;0.000700197;0.000700197;0.000700197;0.000488841;0.000264782;0;0
A_2;3.62052e-05;0.000114814;0.000229225;0.00039447;0.000706437;0.00109931;0.00184489;0.00275296;0.00395164;0.00437528;0.00440057;0.00444726;0.0046156;0.00229778;0.00229778;0.00229778;0.00160785;0.000873259;0;0
A_3;6.5358e-05;0.000229225;0.000508878;0.000924219;0.00175717;0.00281323;0.00496666;0.00757395;0.0106558;0.0116963;0.0117524;0.0118769;0.0122887;0.00552015;0.00552015;0.00552015;0.00356421;0.00147568;0;0
C_4;0.000125994;0.00039447;0.000924219;0.00202415;0.00404467;0.00714106;0.0130565;0.0190823;0.0252285;0.0267963;0.0268554;0.0269861;0.0249678;0.0181993;0.00847111;0.0061239;0.00416796;0.00207943;0.000603754;0.000139873
A_5;0.000196552;0.000706437;0.00175717;0.00404467;0.00820796;0.013931;0.0259249;0.0418595;0.0596996;0.0666565;0.0668406;0.0675258;0.067224;0.0307559;0.0210277;0.0186805;0.00878539;0.00352443;0.000603754;0.000139873
C_6;0.000328442;0.00109931;0.00281323;0.00714106;0.013931;0.0247509;0.0449983;0.069822;0.101313;0.110441;0.110632;0.111336;0.0991588;0.0626907;0.0334127;0.0198717;0.00997658;0.00471562;0.00179495;0.000446343
C_7;0.000553431;0.00184489;0.00496666;0.0130565;0.0259249;0.0449983;0.0786128;0.119909;0.175115;0.191833;0.192042;0.192809;0.159914;0.123446;0.0559382;0.0212691;0.011374;0.00611305;0.00319238;0.000889846
C_8;0.000824511;0.00275296;0.00757395;0.0190823;0.0418595;0.069822;0.119909;0.184088;0.278008;0.309829;0.310077;0.310961;0.24368;0.207212;0.0711069;0.0228361;0.012941;0.00768002;0.00475935;0.00121992
C_9;0.00120974;0.00395164;0.0106558;0.0252301;0.0597011;0.101316;0.175118;0.278011;0.442224;0.540951;0.541838;0.548287;0.314152;0.27952;0.134106;0.0307786;0.0390998;0.134187;0.131266;0.127646
C_10;0.00133627;0.00437528;0.0116963;0.0267979;0.066658;0.110445;0.191836;0.309832;0.540951;0.639678;0.640565;0.549795;0.314804;0.280171;0.134243;0.0307985;0.0391198;0.134207;0.131286;0.127653
G_11;0.00134247;0.00440057;0.0117524;0.026857;0.0668422;0.110635;0.192045;0.310081;0.541838;0.640565;0.641452;0.550681;0.31569;0.280274;0.134346;0.0309011;0.0391607;0.134221;0.131286;0.127653
G_12;0.00135501;0.00444726;0.0118769;0.0269877;0.0675273;0.111339;0.192813;0.310965;0.548287;0.549795;0.550681;0.459911;0.244312;0.207938;0.0838498;0.0269663;0.0368367;0.0322818;0.0296791;0.0269458
U_13;0.00140472;0.0046156;0.0122887;0.0249678;0.067224;0.0991588;0.159914;0.24368;0.314149;0.3148;0.315687;0.244309;0.149036;0.12523;0.0603534;0.0204676;0.0169076;0.0134034;0.0115843;0.00969977
G_14;0.000700197;0.00229778;0.00552015;0.0181993;0.0307559;0.0626907;0.123446;0.207212;0.279517;0.280168;0.280271;0.207934;0.12523;0.0989915;0.0415884;0.013814;0.0112185;0.00810799;0.00642706;0.00506415
G_15;0.000700197;0.00229778;0.00552015;0.00847111;0.0210277;0.0334127;0.0559382;0.0711069;0.134103;0.13424;0.134343;0.0838467;0.0603534;0.0415884;0.0198644;0.0078437;0.00499886;0.00303726;0.00193873;0.00115273
U_16;0.000700197;0.00229778;0.00552015;0.0061239;0.0186805;0.0198717;0.0212691;0.0228361;0.0307754;0.0307954;0.030898;0.0269632;0.0204676;0.013814;0.0078437;0.00372948;0.00238196;0.0014955;0.000994685;0.000636301
U_17;0.000488841;0.00160785;0.00356421;0.00416796;0.00878539;0.00997658;0.011374;0.012941;0.0390967;0.0391166;0.0391576;0.0368336;0.0169076;0.0112185;0.00499886;0.00238196;0.00153295;0.000941024;0.000584357;0.000391474
U_18;0.000264782;0.000873259;0.00147568;0.00207943;0.00352443;0.00471562;0.00611305;0.00768002;0.134184;0.134204;0.134217;0.0322786;0.0134034;0.00810799;0.00303726;0.0014955;0.000941024;0.000539355;0.000275861;0.000181808
G_19;0;0;0;0.000603754;0.000603754;0.00179495;0.00319238;0.00475935;0.131263;0.131283;0.131283;0.0296759;0.0115843;0.00642706;0.00193873;0.000994685;0.000584357;0.000275861;6.54789e-05;2.81631e-05
G_20;0;0;0;0.000139873;0.000139873;0.000446343;0.000889846;0.00121992;0.127643;0.12765;0.12765;0.0269426;0.00969977;0.00506415;0.00115273;0.000636301;0.000391474;0.000181808;2.81631e-05;5.22605e-06
This data can be visualized in heatmaps as discussed for the minimal energy heatmap.
Accessibility and unpaired probabilities
Accessibility describes the availability of an RNA subsequence for intermolecular base pairing. It can be expressed in terms of the probability of the subsequence to be unpaired (its unpaired probability Pu).
A limited accessibility, i.e. a low unpaired probability, can be incorporated into the RNA-RNA interaction prediction by adding according energy penalties. These so called ED values are transformed unpaired probabilities, i.e. the penalty for a subsequence partaking in an interaction is given by ED=-RT log(Pu), where Pu denotes the unpaired probability of the subsequence. Within the IntaRNA energy model, ED values for both interacting subsequences are considered.
To globally turn off accessibility consideration, set --acc=N.
Accessibility incorporation can be disabled separately for query or target sequences using
--qAcc=N or --tAcc=N, respectively.
A setup of --acc=C (default, as for --qAcc=C and --tAcc=C) enables accessibility computation
using the selected energy model for query or target sequences, respectively.
Using --accNoLP and --accNoGUend, the consideration of lonely base pairs
and GU-helix ends can be disabled for accessibility computation (for default
energy model V).
Local versus global unpaired probabilities
Exact computation of unpaired probabilities (Pu terms) is considers all possible structures the sequence can adopt (the whole structure ensemble). This is referred to as global unpaired probabilities as computed e.g. by RNAup.
Since global probability computation is (a) computationally demanding and (b) not reasonable for long sequences, local RNA folding was suggested, which also enables according local unpaired probability computation, as e.g. done by RNAplfold. Here, a folding window of a defined length ‘screens’ along the RNA and computes unpaired probabilities within the window (while only intramolecular base pairs within the window are considered).
IntaRNA enables both global as well as local unpaired probability computation.
To this end, the sliding window length --accW and the maximal base pair span
--accL (<= --accW) have to be specified in order to enable/disable
local folding.
Use case examples global/local unpaired probability computation
The use of global or local accessibilities can be defined independently
for query and target sequences using --qAccW|L and --tAccW|L, respectively.
Here, --?AccW defines the sliding window length (0 sets it to the whole sequence length)
and --?AccL defines the maximal length of considered intramolecular base pairs,
i.e. the maximal number of positions enclosed by a base pair
(0 sets it to the whole sequence length). Both can be defined
independently while respecting AccL <= AccW.
# using global accessibilities for target and query
IntaRNA [..] --accW=0 --accL=0
# using global accessibilities for query and local ones for target
IntaRNA [..] --qAccW=0 --qAccL=0 --tAccW=150 --qAccL=100
Constraints for accessibility computation
For some RNAs additional accessibility information is available. For instance, it might be known from experiments that some subsequence is unpaired or already bound by some other factor. The first case (unpaired) makes such regions especially interesting for interaction prediction and should result in no ED penalties for these regions. In the second case (blocked) the region should be excluded from interaction prediction.
To incorporate such information, IntaRNA provides the possibility to constrain
the accessibility computation using the --qAccConstr and --tAccConstr
parameters. Both take a string encoding for each sequence position whether it is
.unconstrainedxfor sure accessible (unpaired)ppaired intramolecularly with some other position of this RNAbblocked by some other interaction (implies single-strandedness)
Note, blocked regions are currently assumed to be bound single-stranded by some other factor and thus are treated as unpaired for ED computation.
# constraining some central query positions to be blocked by some other molecules
IntaRNA [..] --query="GGGGGGGCCCCCCC" \
--qAccConstr="...bbbb......."
It is also possible to provide a more compact index-range-based encoding of the constraints, which is especially useful for longer sequences or if you have only a few constrained regions. To this end, one can provide a comma-separated list of index ranges that are prefixed with the according constraint letter from above and a colon. Best check the following examples, which should give a good idea how to use. Note, indexing is supposed to be based on a minimal index of 1 and all positions not covered by the encoding are assumed to be unconstrained (which must not to be encoded explicitely).
# applying the same constraints by different encodings to query and target
# example 1
IntaRNA [..] --qAccConstr="...bbbb....." --tAccConstr="b:4-7"
# example 2
IntaRNA [..] --qAccConstr="..bb..xxp.bb" --tAccConstr="b:3-4,11-12,x:7-8,p:9-9"
When constraining the accessibility computation, all predictions are conditional
predictions for the given accessibility constraints.
If you are still interested in non-conditional results/energies, you will have to
correct the energy computation by providing a respective energy shift to be
applied. By setting --energyAdd=.., all energy evaluations will add your given
term and thus IntaRNA will use and provide corrected energy terms.
Scaling factors for partition function computation for accessibility estimation
To compute the unpaired probabilities, which are the base for accessibility estimation, one needs to calculate so called partition functions of structure ensembles. The value of these numbers can be very large for longer sequences, since the RNA structure space growths exponentially with sequence length. Thus, the respective methods from the Vienna RNA package employ mathematical tricks to scale the partition function values in order to keep them within ranges that can be handled with normal data structures.
If you are investigating very long (or special) sequences, these tricks might be insufficient and the accessibility computation will fail with an error similar to the one shown below.
This error, raised by the underlying routine from the Vienna RNA package, shows that
the value of the partition function Q exceeds the possible range for such numbers.
This problem can be tackled, when supporting the partition function scaling with a
sequence specific scaling factor (the pf_scale mentioned within the error above).
You can set this factor using --qPfScale|--tPfScale for query|target sequence,
respectively. The larger the value, the higher Q values can be dealt with but with the
cost of precision. Too high values will render the accessibility computation useless,
since the precision is so low that all unpaired probabilities are dropping to zero.
Thus, be careful when setting the factor. The
RNAplfold manual
recommends values between 1 and 2. But higher values might be needed for very long
sequences.
Read/write accessibility from/to file or stream
It is possible to read precomputed accessibility values from file or stream to avoid their runtime demanding computation. To this end, we support the following formats
| Input format | produced by |
|---|---|
| RNAplfold unpaired probabilities | RNAplfold -u or IntaRNA --out=*Pu: |
| RNAplfold-styled ED values | IntaRNA --out=*Acc: |
| .. with gzip-compression | IntaRNA --out=*:*.gz |
The RNAplfold format is a table encoding of a banded upper triangular matrix
with band width l. First row contains a header comment on the data starting with
#. Second line encodes the column headers, i.e. the window width per column.
Every successive line starts with the index (starting from 1) of the window end
followed by a tabulator separated list for each windows value in increasing
window length order. That is, column 2 holds values for window length 1, column
3 for length 2, … . The following provides a short output/input
example for a sequence of length 5 with a maximal window length of 3.
#unpaired probabilities
#i$ l=1 2 3
1 0.9949492 NA NA
2 0.9949079 0.9941056 NA
3 0.9554214 0.9518663 0.9511048
4 0.9165814 0.9122866 0.9090283
5 0.998999 0.915609 0.9117766
6 0.8549929 0.8541667 0.8448852
Use case examples for read/write accessibilities and unpaired probabilities
If you have precomputed data, e.g. the file plfold_lunp with unpaired probabilities
computed by RNAplfold, you can run
# fill accessibilities from RNAplfold unpaired probabilities
IntaRNA [..] --qAcc=P --qAccFile=plfold_lunp
# fill accessibilities from RNAplfold unpaired probabilities via pipe
cat plfold_lunp | IntaRNA [..] --qAcc=P --qAccFile=STDIN
Another option is to store the accessibility data computed by IntaRNA for successive calls using
# storing and reusing compressed (target) accessibility (Pu) data for successive IntaRNA calls
IntaRNA [..] --out=tPu:intarna.target.pu.gz
IntaRNA [..] --tAcc=P --tAccFile=intarna.target.pu.gz
# piping (target) accessibilities (ED values) between IntaRNA calls
IntaRNA [..] --out=tAcc:STDOUT | IntaRNA [..] --tAcc=E --tAccFile=STDIN
Note, for multiple sequences in FASTA input, one can also load the
accessibilities (for all sequencces) from file. To this end, the file names
have to be suffixed with with -s#, where # denotes the respective sequence’s number (where indexing
starts with 1) within the FASTA input using a common suffix after the index.
The file name without -s# is to be provided to the according --?AccFile argument.
The files generated by --out=?Acc:... are already conform to this requirement,
such that you can use the use case examples from above also for multi-sequence
FASTA input.
Note, this is not supported for a piped setup (e.g. via --out=tAcc:STDOUT
as shown above), since this does not produce the according output files!
Library for integration in external tools
The IntaRNA package also comes with a C++ library libIntaRNA.a containing the core classes
and functionalities used within the IntaRNA tool. The whole library comes with
an IntaRNA namespace and exhaustive class and member API documentation that is
processed using doxygen to generate html/pdf versions.
When IntaRNA is build while pkg-config is present, according pkg-config
information is generated and installed too.
Mandatory Easylogging++ initalization !
Since IntaRNA makes heavy use of the Easylogging++ library, you have to add (and adapt)
the following code to your central code that includes the main() function:
// get central IntaRNA-lib definitions and includes
#include <IntaRNA/general.h>
// initialize logging for binary
INITIALIZE_EASYLOGGINGPP
[...]
int main(int argc, char **argv){
[...]
// set overall logging style
el::Loggers::reconfigureAllLoggers(el::ConfigurationType::Format, std::string("# %level : %msg"));
// no log file output
el::Loggers::reconfigureAllLoggers(el::ConfigurationType::ToFile, std::string("false"));
el::Loggers::reconfigureAllLoggers(el::ConfigurationType::ToStandardOutput, std::string("true"));
// set additional logging flags
el::Loggers::addFlag(el::LoggingFlag::DisableApplicationAbortOnFatalLog);
el::Loggers::addFlag(el::LoggingFlag::LogDetailedCrashReason);
el::Loggers::addFlag(el::LoggingFlag::AllowVerboseIfModuleNotSpecified);
// setup logging with given parameters
START_EASYLOGGINGPP(argc, argv);
[...]
}
Note further, to get the library correctly working the following compiler flags are used within the IntaRNA configuration:
CXXFLAGS=" -DELPP_FEATURE_PERFORMANCE_TRACKING -DELPP_NO_DEFAULT_LOG_FILE "
Attributions
![]()
![]()
![]() Designed by Freepik
Designed by Freepik